Fabio Pietrucci, Sorbonne Université, IMPMC, Paris
fabio.pietrucci@sorbonne-universite.fr
Molecular dynamics simulations can complement experiments by providing detailed, atomic- scale information about transition mechanisms between different states of materials, including nanostructures, solids, solutions, biomolecules etc. If interatomic forces are accurately described, in principle, transition states (difficult to capture in experiments due to their short lifetime) can be identified, barriers and rates can be quantitatively estimated. This kind of information can be useful to characterize the behavior of materials in real conditions of temperature and pressure, and to make sense of synthesis or degradation processes.
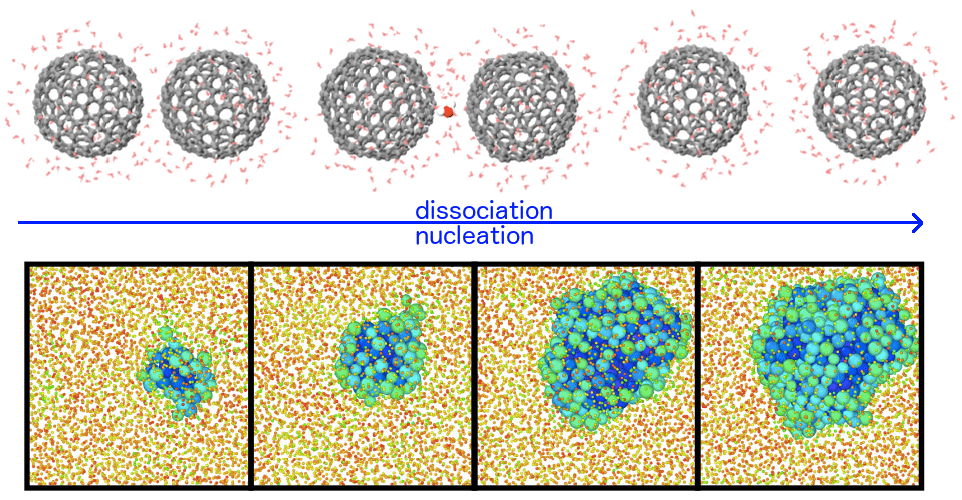
However, a major hurdle consists in the long characteristic timescale of many transformation processes, exceeding by far what can be simulated today (typically, from nanoseconds to microseconds). I will present some methods developed in my group, that tackle the latter challenge exploiting two strategies. The first consists in applying external forces on some flexible order parameters, specifically designed to capture and accelerate changes in the topology of the atomic network during a transformation. The second consists in directly exploring transition states and mechanisms using « transition path sampling » techniques: the resulting trajectories, projected on an order parameter, can be effectively modeled by Langevin equations, that in turn allow (based on a recently demonstrated variational principle) to optimize in a unified way the order parameter definition, the free-energy landscape and the kinetic rate. I will discuss applications to problems ranging from structural changes in core-shell nanoparticles, to crystal nucleation, to protein-protein interaction.
References
F. Pietrucci, Rev. Phys. 2, 32 (2017).
S. Pipolo, M. Salanne, G. Ferlat, S. Klotz, A.M. Saitta, F. Pietrucci, Phys. Rev. Lett. 119, 245701 (2017). L. Mouaffac, K. Palacio-Rodriguez, F. Pietrucci, J. Chem. Theory Comput. 19, 5701 (2023).