Résumé
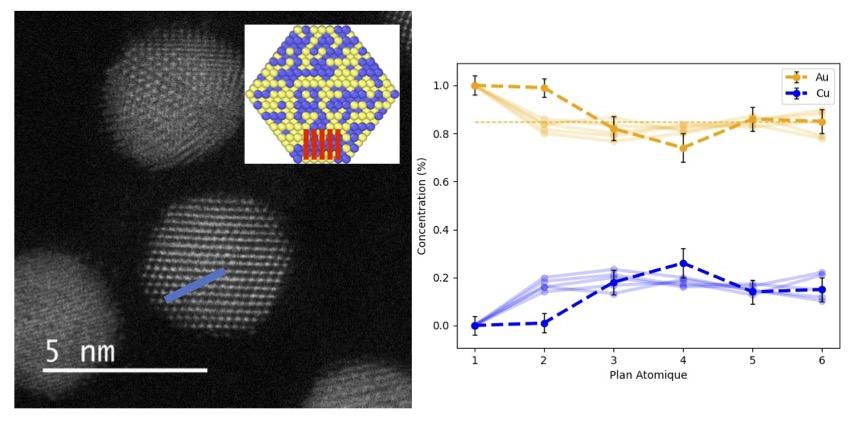
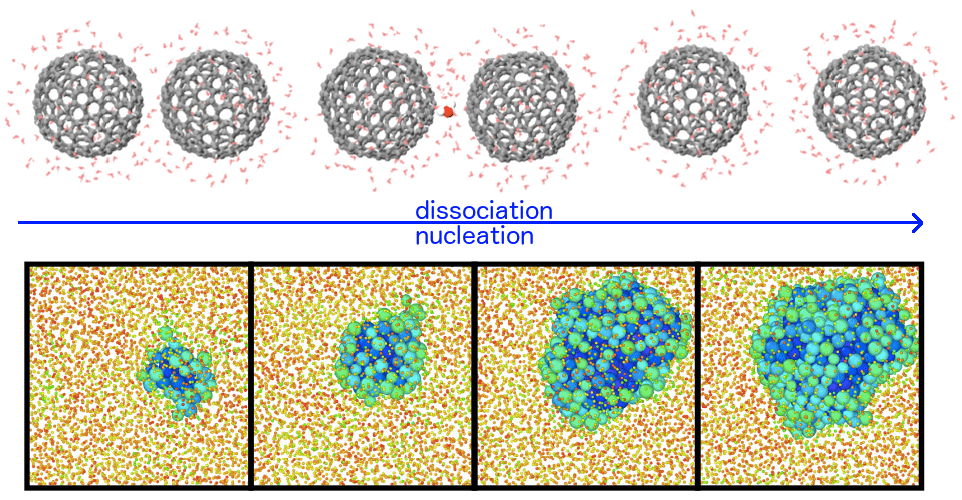
Les nanoparticules métalliques (NP) possèdent des propriétés uniques, distinctes des matériaux massifs, offrant ainsi des applications potentielles dans divers domaines tels que la mécanique, la catalyse ou encore l’optique. Dans ce contexte, cette thèse étudie comment les propriétés mécaniques des NP, influencées par la forme, la taille et la composition, affectent leurs propriétés électroniques. En couplant des calculs de type dynamique moléculaire et des simulations par éléments finis, nous démontrons l’effet significatif de la forme sur la réponse élastique effective. Nos résultats soulignent que la plasticité est contrôlée à la fois par la forme et la taille, avec un effet de taille universel pour les NP cristallines cubiques à faces centrées. Dans le cas de NPs d’alliages, des mécanismes de renforcement et d’adoucissement sont observés, indiquant l’influence de l’ordre local sur l’élasticité et la plasticité. Enfin, grâce à un modèle reposant sur un formalisme de type liaisons fortes et des calculs ab initio, nous révélons que la déformation plastique crée de nouveaux sites réactifs à la surface des NP.
Candidat :
Matteo Erbi’
Jury :
Pr. Riccardo Ferrando – Université de Gênes (Italie)- Rapporteur
Dr. Julien Godet – Université de Poitiers – Rapporteur
Pr. Francesco Montalenti – Université de Milan-Bicocca (Italie) – Examinateur
Dr. Christine Mottet – CINaM – Examinatrice
Dr. Fabio Pietrucci – Sorbonne Université – Examinateur
Dr. Barbaru Putz- Empa (Suisse) – Examinatrice
Dr. Riccardo Gatti – LEM – Co-Directeur de thèse
Dr. Hakim Amara – LEM – Directeur de thèse
Vendredi 24 novembre 2023 à 14h00
Salle Contensou, ONERA, 29 Avenue de la Division Leclerc,92320, Chatillôn