
Speaker: Dr. Moustafa Benyoucef
Date and Location: Tuesday 10/12/19, 10h30 LEM meeting room (E2.01.20), Châtillon.

Speaker: Dr. Moustafa Benyoucef
Date and Location: Tuesday 10/12/19, 10h30 LEM meeting room (E2.01.20), Châtillon.

Fig. 1 (top) Experimental reconstruction of the u111 displacement field on a 250 nm Pt NP (bottom) u111 displacement field obtained by energy minimization of a simulated Pt NP (right) εxx, εyy and εzz components of the strain tensor derived from the simulation
Physical properties at small length scale deviate strongly from the bulk counterpart, typically below the micrometer. For instance, mechanical strength increases with reducing size, large residual strain due to processing are present in nanostructures. Thus a better understanding of the physical properties in relationship with the microstructure is needed for nanoscale materials. Because of its good spatial resolution (~ 10 nm) and excellent sensitivity to atomic displacements and local strain [1,2], Bragg coherent diffraction imaging (BCDI) has emerged in the past two decades as a powerful tool to probe the structure and local displacement field inside nanoscale objects [3]. When combined with in situ mechanical loading, BCDI is particularly relevant for the study of defect nucleations in isolated nanoparticles [4] or to investigate intragranular deformation mechanisms in polycrystalline thin films [5].
Nowadays, the length scales that are accessible by BCDI and that can be simulated by Molecular Dynamics (MD) simulation are almost converging. The coupling between the two methods is therefore particularly relevant and allows to get a detailed picture of the deformation mechanisms in nanostructures at the atomic scale. This coupled approach has been used to study the surface relaxation of metallic nanoparticles (Au, Pt). An excellent quantitative agreement is obtained between the component of the displacement field measured experimentally and calculated by energy minimization (Molecular Statics) (Fig. 1). With this approach, the measurement of only one Bragg reflection is required to derive the 3D displacement field and the six independent components of the strain tensor from the simulation [6]. The two techniques can also be combined to identify defect structures nucleated during in situ mechanical loading [4,5] and to interpret the evolution of the strain field in nanoparticle catalysts during gas reaction [7,8]..
[1] Watari, M. et al. Nature Materials 10, 862–866 (2011).
[2] Labat, S. et al. ACS Nano 9, 9210–9216 (2015).
[3] Robinson, I. & Harder, R. Nat Mater 8, 291–298 (2009).
[4] Dupraz, M. et al. Nano Lett. 17(11) (2017).
[5] Cherukara, M. et al. Nat. Comm. 9 (2018).
[6] Dupraz et al. to be submitted (2019)
[7] Kim, D. et al. Nat. Comm. 9, 3422 (2018).
[8] Dupraz, M. et al. in preparation
Speaker: Dr. Maxime Dupraz
Date and Location: Monday 25/11/19, 14h00 LEM meeting room (E2.01.20), Châtillon.
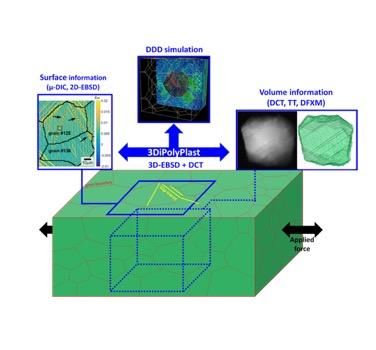
Understanding the deformation processes that lead to the failure of polycrystalline structural materials is one of the main challenges in materials science. Significant progress has been made in recent decades, thanks to the development of new experimental characterisation techniques and advanced simulation methods.
However, the localisation of plasticity in slip bands and the propagation of plasticity through a polycrystalline aggregate are not fully understood.
One of the difficulties in modelling the mechanical behaviour of polycrystalline materials is its intrinsically multi-scale nature. Inelastic deformation mechanisms occur at the dislocation scale (dislocation is a crystalline defect ,vector of plastic deformation) that result in the formation of intra-granular microstructures which, in turn, interact with grain boundaries.
This internship project aims to study the first stages of plastic deformation in policrystalline materials (Cu, Ni) using dislocation dynamics (DD). In particular, the microMegas DD code coupled with a mechanical solver will be used, to correctly handle boundary conditions, to capture the onset of plastic deformation localisation and thus model the interaction between a slip band and grain boundaries.
This mechanism will be studied in a “model” polycrystalline aggregate, and then, if possible, extended to a digital twin of a real polycrystal.
Job: Internship (4-6 months)
Academic level : Master degree
Location: LEM, Châtillon
Expertise: : Solid state physics, Materials Science. Interest in theoretical physics and numerical simulations
Contacts: Benoit Devincre

The proposed internship topic is part of a cooperative project on the development of new catalysts (metal or bimetallic nanoparticles) for the growth of carbon nanotubes, aiming at controlling their electronic properties during their synthesis.
The nanoparticles as catalysts will be synthesized, in collaboration with the partnering group of Pr V. Huc at the Institute of Molecular Chemistry and Orsay Materials (ICCMO) by combining surface chemistry and coordination chemistry. We propose to compare the same set of metallic or bimetallic nanoparticles synthesized using very different synthetic pathways and then we will compare the catalytic action of these nanoparticles in the growth of nanotubes.
We will focus on the Ni-Ru catalytic system, identified as a good candidate for achieving the desired electronic character selectivity.
– A first part of the experimental work will be to synthesize the nanoparticles using soft chemistry via the colloidal route (optimization of the synthesis by first reducing the Ru3 + in Ru2 + and then combining it with Ni) and Prussian Blue Analogues at ICMMO. The latter will be calcined under argon in order to obtain carbureted species. We will do the structural study (size distribution, composition, crystalline structure) at LEM. For the latter, we will use a powerful set of investigation techniques (high-resolution transmission electron microscopy, diffraction, energy loss spectroscopy and X-ray spectroscopy) present in the laboratory (LEM).
– A second part of the experimental work will be to grow carbon nanotubes by CVD using the nanoparticles developed as a catalyst (colloidal, by “Prussian blue” and Prussian blue “carbides”) and to characterize their structures (chirality, length, type of adhesion to the nanoparticle) by TEM (imaging and diffraction) and Raman spectroscopy. These three sets of catalysts should be compared to observe the influence of the synthesis route and the influence of the carburized phase on the growth of carbon nanotubes.
Job: Internship (4-6 months)
Academic level : Master degree
Location: LEM, Châtillon
Expertise: Good training in condensed matter physics and chemistry with a major focus on nanoscience and courses on synthesis and characterization.
Strong interest for experiments
Contacts: Armelle Girard, Annick.Loiseau

The proposed internship topic is part of a cooperative project on the development of new bimetallic catalysts in the solid solution state for the growth of carbon nanotubes, aiming at controlling their electronic properties during their synthesis.
Although several techniques are available for the synthesis of transition metal-based bimetallic catalysts, they generally lead to nanoparticles with a core/shell or janus morphology. Nevertheless, our previous studies have shown that it is possible to synthesize bimetallic particles in solid solution state, that is with no elemental segregation within the nanoparticle by combining surface chemistry and coordination chemistry under particular temperature conditions.
However, studying the thermodynamic behavior of these bimetallic catalysts often requires the implementation of complicated experimental techniques that often need to be coupled with theoretical approaches, even if the latter are still far from being able to reach such a level of complexity while remaining predictive. In collaboration with the partnering group of numerical simulations at the Institute of Molecular Chemistry and Orsay Materials (ICCMO) (Jérôme Creuze and Fabienne Berthier), we will undertake theoretical study to better characterize the behavior of these nanoparticles, in equilibrium and under ultra-vacuum first. Indeed, it is necessary to know the thermodynamics of nanoalloys as isolated systems before studying the influence of external perturbations. We will also study the kinetics of return to equilibrium in order to determine the stability and the lifetime of metastable configurations that will have been identified during the first step of the study.
The internship will thus identify and quantify the key thermodynamic parameters involved in the distribution of constituents within the bimetallic nanoparticles and understand how the thermodynamic variables influence the equilibrium configuration using these parameters. The candidate will be able to compare his results with experiences when possible.
Job: Internship (4-6 months)
Academic level : Master degree
Location: LEM, Châtillon
Expertise: Good training in condensed matter physics and chemistry with a major focus on nanoscience, thermodynamics. Strong interest for numerical calculations
Contacts: Armelle Girard, Annick.Loiseau

Diffusion processes in solids are relevant for the kinetics of many microstructural changes that occur during preparation, processing, and heat treatment of materials. Typical examples are nucleation of new phases, diffusive phase transformations, precipitation of a second phase, recrystallisation, high-temperature creep, and thermal oxidation. To reach a deep understanding of diffusion in solids, one needs information on the position of atoms and how they move in solids. The atomic mechanisms of diffusion in crystalline solids are closely connected with defects. Point defects such as vacancies or interstitials are the simplest defects and often mediate diffusion in crystals. Ab initio methods as DFT (Density Functional Theory) can provide fundamental information, such as the stable positions of atoms in a crystal lattice and their jumping rates between two neighour sites. However, it is not trivial to obtain diffusion coefficients from these fundamental properties because, in complex solid crystals, there are usually various point defects (vacancy and interstitial positions) and hence several diffusion paths are possible for the diffusing atom. Analytical solutions of multi-state diffusion problems are generally complex. A good alternative is to resource to Kinetic Monte Carlo (KMC), which is a particular Monte Carlo method used for processes with known rates such as atom migration. It consists of mapping N possible events that can occur from a given state. Each event is defined by jump frequency, displacement and cumulative function of the jump frequency: all these input quantites can be obtained from the DFT calculations.
The main aim of this project is to develop a Kinetic Monte Carlo code for studying diffusion of atoms in crystals. The code will receive as input data the results of DFT calculations, which will be performed with the VASP code. The first applications will address the diffusion of interstitial atoms (B, O, N, C) in Ti-Al alloys. The code will be validated by comparison with the analytical solution of the simplest crystal lattices (as the tetragonal TiAl) and then it will be applied for the study of diffusion in more complex geometries (as the hexagonal Ti3Al).

The control of the composition and morphology of materials at the nanoscale allowed to disclose novel structural, electronic and chemical properties which are fundamental for many recent technological advances. Amongst nanostructures, 2D materials are a class formed of materials which cryistallise as atomically-thin layers. Since the discovery of graphene in the early 2000s, the family of 2D materials grew larger, with the emergence of new systems alike the hexagonal boron nitride (hBN) or the black phosphorus (BP).
Because of their extreme thinness, 2D materials often display electronic properties sizeably different from those of their bulk equivalent. Moreover, their characteristics are strongly influenced by the interaction with the near surroundings: for instance by modifications of the substrate, or changes of their thickness. Van der Waals heterostructures are based on this principle. They are built by stacking layers of different 2D materials on top of each other, so that several properties are combined in the same system and tuned in a controlled fashion. This allows to engineer specific properties aimed for technological development or fundamental research.
In this context, we will consider heterostructures based on hBN and/or BP layers. In order to study these systems from a theoretical perspective, we will elaborate a mixed approach combining analytical and numerical developments in the tight-binding formalism, with ab-initio simulations. The latter will be done on simple reference systems, with the intent to establish a quantitative basis for the parametrization of tight-binding models. This will make possible the investigation of extended systems like realistic heterostructures. More precisely, the objectif will be that of studying the influence of the environment (substrate, stacking …) on the electronic and optical properties of van der Waals heterostructures based on hBN and BP.
Another specificity of this work will consist on coupling the theoretical study with diverse experimental techniques, namely thanks to our rich collaboration network.

Georges Saada was born on August 10, 1932 in Sfax, Tunisia. He left Tunisia at the age of seventeen, after high school and obtaining his baccalaureate. In Paris, he prepared the Grandes Ecoles at the Louis-le-Grand and Buffon high schools. He joined the École Polytechnique in 1952 and then prepared a thesis in metal physics. He also graduated at the École Nationale Supérieure des Télécommunications.
After military service, his career began in the army, which he left in 1960. Indeed his skills led him to scientific research. After five years at the Institut de Recherche de la Sidérurgie, as a research engineer, he chose to focus his career on university teaching. First a lecturer at the University of Lille, he participated in 1969 in the creation of the University of Paris XIII Villetaneuse where he became a professor in 1971. From 1973 to 1981, he was at the head of the Laboratoire des Propriétés Mécaniques et Thermodynamiques des Matériaux.
In 1981, he was appointed Head of Mission for Higher Education at the Ministry of National Education, Alain Savary.
He returned to the University of Paris XIII and joined the Laboratoire d’Etude des Microstructures in 1990.
Georges Saada played a pioneering role in the field of plasticity of materials. His work has had a major impact on the development of this discipline, with seminal contributions to the understanding of the physical mechanisms that cause deformation of metal alloys. His work has been recognized by the award of the Grande Médaille de la Société Française de Métallurgie et de Matériaux in 2008.

Congratulations to Viviane Cothias-Laut (our assistant) has been selected to publish her work in the art magazine Art Folio.
Here is a glimpse of her creations:


Phd candidate: Maoyuan JIANG
Directeur de thèse : Benoit DEVINCRE
Co-directeur de thèse : Ghiath MONNET
Dislocation Dynamics (DD) simulations are used to investigate the Hall-Petch (HP) effect and back stresses induced by grain boundaries (GB) in polycrystalline materials.
The HP effect is successfully reproduced with DD simulations in simple periodic polycrystalline aggregates composed of 1 or 4 grains. In addition, the influence of grain shape was explored by simulating grains with different aspect ratios. A generalized HP law is proposed to quantify the influence of the grain morphology by defining an effective grain size. The average value of the HP constant K calculated with different crystal orientations at low strain is close to the experimental values.
The dislocations stored during deformation are mainly located at GB and can be dealt with as a surface distribution of Geometrically Necessary Dislocations (GNDs). We used DD simulations to compute the back stresses induced by finite dislocation walls of different height, width, density and character. In all cases, back stresses are found proportional to the surface density and their spatial variations can be captured using a set of simple empirical equations. The back stress calculation inside grains is achieved by adding the contributions of GNDs accumulated at each GB facet.
These back stresses are found to increase linearly with plastic strain and are independent of the grain size. The observed size effect in DD simulations is attributed to the threshold of plastic deformation, controlled by two competing mechanisms: the activation of dislocation sources and forest strengthening. Due to strain localization in coarse-grained materials, the pile-up model is used to predict the critical stress. By superposing such property to the analysis we made from DD simulations in the case of homogeneous deformation, the HP effect is justified for a wide range of grain sizes.
Tuesday 04/05/2019, 13h30
Amphi 3, Bâtiment Eiffel, CentraleSupélec, 8-10 rue Joliot-Curie, 91190 Gif-sur-Yvette