Abstract
The analysis of nanoparticles (NPs) on a nanometric scale for applications in real-life conditions remains a considerable challenge at the present time. In this context, the use of bi-metallic NPs is strongly envisaged in the field of catalysis, with the function of promoting and accelerating the kinetics of surface chemical reactions. It is therefore essential to describe the structure and chemical composition of the surfaces, which interact directly with the surrounding medium in which the NPs are immersed. In the context of this thesis, we have developed a combined theoretical and experimental approach at the atomic scale, with the aim of studying two types of alloy in particular: Gold-Copper (Aux-Cu1-x) and Nickel-Aluminium (Nix-Al1-x).
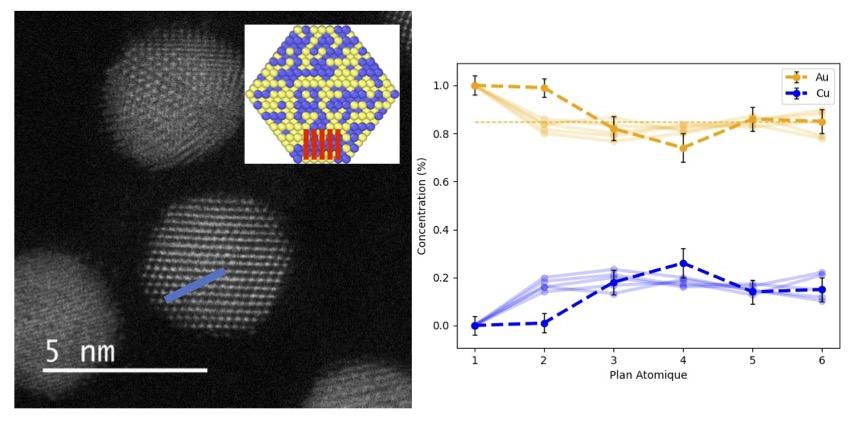
Using laser synthesis of 5 nm facetted octahedral Aux-Cu1-x NPs and aberration-corrected electron microscopy observations in probe mode, we developed a method for analyzing the chemical composition of each atomic plane. In this way, we have demonstrated a strong segregation effect of gold on the surface, as well as different concentration profiles within the NPs depending on the chemical order (ordered or disordered). In the case of an ordered Au0.5Cu0.5 composition of L10 phase, we have characterized a structure rarely observed until now, corresponding to the presence of the three possible variants of L10 phase within the same particle. In parallel, atomic-scale simulations have enabled more precise analyses to be carried out, considering infinite plane stacks and NPs of different sizes and compositions.
The excellent agreement between simulations and experimental analyses strengthens the relevance of our results and demonstrates the importance of this dual approach, which we subsequently applied to the study of the surface properties of Nix-Al1-x-type NPs. First, we optimized the synthesis parameters to obtain NPs with defined sizes and compositions. Experimental surface analyses coupled with atomistic simulations enabled us to observe a hitherto unseen phenomenon. Indeed, an almost complete segregation of the aluminum appears until the formation of NPs adopting a core (Nickel) – shell (Aluminum) structure, for all the concentrations studied, thus preventing any alloy formation. This is all the more surprising given that, in the bulk state and for a composition of 50% nickel and 50% aluminium, the ordered B2 phase, known for its stability and resistance to corrosion, appears. These striking structural differences between the nanometric and macroscopic scales once again demonstrate the unique physics that exist in the world of the infinitely small.
PhD Candidate :
Grégoire Breyton
Jury :
Dr. Christine Goyhenex – IPCMS – Referee
Pr. Claude Henry – CINaM – Referee
Dr. Pascale Bayle – CEA/Grenoble – Reviewer
Dr. Geoffroy Prévot – INSP – Reviewer
Dr. Hakim Amara – LEM – PhD co-supervisor
Pr. Christian Ricolleau – MPQ – PhD supervisor
Friday 15th December 2023 at 14h00
Pierre-Gilles de Genes Amphitheater, Paris Cité University, Paris